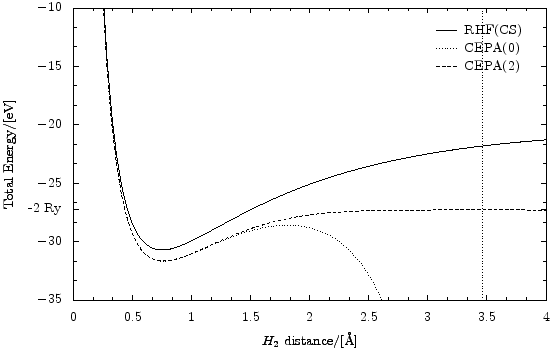
The following script produces this picture (as eps):
It does this by repeatedly doing standard RHF/CEPA calculations on dynamically created versions of the H2 molecule (for all given distances) and then using the PyX vector graphics library to paint a diagram with the so aquired data. What we can see here is that it would be very easy to modify this script to do something different if that would be wished. Only minimal human interaction is required to build graphs in this fashion.
"""This script useses the PyX graphics library (http://pyx.sourceforge.net/) to paint a potential energy surface of the H_2 dissociation. The result shows that RHF cannot handle the dissociation since the surpression of charge fluctuations becomes important (i.e., 'static correlations' get strong.). In RHF and CEPA(0) the H_2 molecule does not dissociate in two H atoms. CEPA(2), however, can handle this test.""" from ct8k import * from ct8kpy import * from pyx import * MainDir = "../" for Basis in ["STO-3G","cc-pVDZ","cc-pVTZ"]: LoadBasisLibrary( MainDir + "basis_lib/%s.libmol" % Basis ) Basis = "cc-pVDZ" SetOutputUnits( ENERGY_ElectronVolts, DISTANCE_Angstrom ) MethodNames = ["RHF(CS)", "CEPA(0)", "CEPA(2)"] # [i]: style of the data lines in which graph for MethodNames[i] is drawn MethodLineStyles = [graph.style.line(), graph.style.line(lineattrs = [style.linestyle.dotted]), graph.style.line(lineattrs = [style.linestyle.dashed])] def CalcH2Energy( Distance ): """returns a list of total energies in the standard output unit for dimolecular distance 'Distance' in atomic units. The list contains the energy values for the methods in 'MethodNames'.""" # dynamically create a new atom set object each time. Atoms = FAtomSet() Atoms.AddAtom( FAtom( FVector3( -Distance/2.0, 0, 0 ), "H", Basis ) ) Atoms.AddAtom( FAtom( FVector3( +Distance/2.0, 0, 0 ), "H", Basis ) ) # do the HF and CEPA calculations.. (Converged, EnergyHf, HfResult, HfParams) = HartreeFock( Atoms ); assert( Converged ) (Converged, EnergyCepa0) = Cepa( HfResult, HfParams.pIntegrator, Method = METHOD_Cepa0, EnergyThreshold = 1e-6 )[:2] assert( Converged ) (Converged, EnergyCepa2) = Cepa( HfResult, HfParams.pIntegrator, Method = METHOD_Cepa2, EnergyThreshold = 1e-6 )[:2] assert( Converged ) # explicitly delete HfParams because it contains the integrator object # which can contain large data sets. The object would be deleted sooner # or later anyway by the garbage collector, but this way seems better. del HfParams # convert energy from atomic units (Hartree) into the standard output # unit (here: electron volts) return map( ConvertEnergy, [EnergyHf, EnergyCepa0, EnergyCepa2] ) def arangeX(xmin,xmax,NumSteps): return map( lambda x: xmin + x*(xmax-xmin)/float(NumSteps-1), range(NumSteps) ) DistancesAu = arangeX( 0.3, 8.0, 100 ) Distances = map( ConvertDistance, DistancesAu ) Energies = map( CalcH2Energy, DistancesAu ) print "H_2 total energy in terms of distance:" for i in range(len(Distances)): print Distances[i], "->", Energies[i] # setup the graph using PyX. On the y-axis, insert a tick for -2 rydbergs, which # is the energy the system is supposed to approach in the infinite distance # limit (electronic energy of two separate H-atoms). g = graph.graphxy( width = 12, x = graph.axis.linear(min=0.0, max=4.0, title="$H_2$ distance/[\\AA]", parter = graph.axis.parter.linear(tickdists=["1/2","1/6"]) ), y = graph.axis.linear(min=-35.0, max=-10.0, manualticks=[ graph.axis.tick.tick(-27.2113845, label="-2 Ry", labelattrs=[] ) ], # text.mathmode title="Total Energy/[%s]" % EnergyUnit(), parter = graph.axis.parter.linear(tickdists=["5/1","5/3"]) ), key = graph.key.key(pos="tr", dist=0.1) ) # transpose the Energy list. From list of [energies for all methods] to # [list of energies] for all methods. Energies = apply(zip, Energies) for (MethodName, MethodLineStyle, MethodEnergyList) in \ zip(MethodNames, MethodLineStyles, Energies): # actually do the plotting. g.plot( graph.data.list( zip(Distances,MethodEnergyList), x=1, y=2, title=MethodName ), styles=[MethodLineStyle] ) g.writeEPSfile(MainDir + "results/H2dissoc.eps")